
Asthma
James Alday
Common Knowledge - A Brief Overview of Asthma
Easily recognizable by symptoms such as coughing, wheezing, and shortness of breath, asthma is a chronic disease of the lung airways affecting millions of people worldwide. Symptom severity and triggers vary from case to case: some people may encounter asthma attacks only during exercise while others may be adversely affected from airborne particulates such as pollen. However, most forms of asthma boil down to the abnormal fashion in how the lung airways are structured and their reaction to stressors.
Around the 19th and early 20th centuries, bronchospasms of the airway smooth muscle were thought to be the primary cause of asthma; appropriately, treatment focus was placed in the development of bronchodilators [1]. But while it was understood that the lungs of an asthmatic were inherently more sensitive than that of a normal individual, the reasoning for such had yet to be discovered. By the 1980’s, the focus of the asthma pathological model shifted from muscle spasms to acute immune response.

Asthma Attack
United States-National Institute of Health: National Heart, Lung, Blood Institute (2013). Asthma attack [illustration]. Retrieved from http://www.nhlbi.nih.gov/health/health-topics/topics/asthma/
It is now understood that both inflammation and the subsequent remodeling of airway epithelia are prominent contributors to the increased sensitivity of the lungs [1]. Inflammation in the lungs is typically the result of the body’s immune response towards an inhaled antigen, with cytokines being released to recruit and activate more immune cells for various allergy signaling cascades. Due to a combination of genetic susceptibility (i.e. atopy), repeated exposure to allergens, and chronic inflammation, the airway epithelia becomes thicker – worsening the effects of any subsequent asthma attack. However, with the identification of immune signaling pathways directly involved in asthma attacks, more potential targets of treatment are being sought after.
Symptoms
-
wheezing
-
persistent coughing
-
shortness of breath
-
feeling of tightness or pain in chest
-
over-production of mucus, leading to excess sputum
Signaling Pathways – Molecular Factors Responsible for Persistence of Asthma
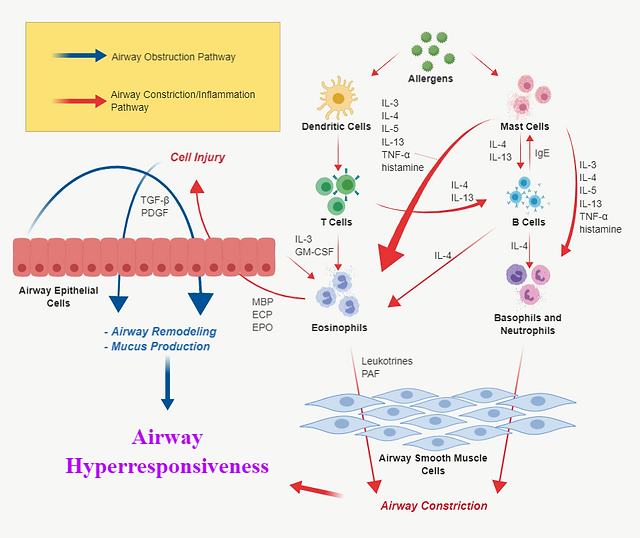
Asthma function can be best described as the synergistic result of signaling pathways that govern airway obstruction and constriction/inflammation (Figure 1). Airway obstruction entails the remodeling of airway epithelia and excess mucus production; airway constriction is the tightening of airway walls and accompanying inflammation that occurs during asthmatic response.
Many of the signaling molecules involved in asthmatic function aren’t mutually exclusive; signaling molecules from airway obstruction pathways either directly or indirectly influence pathways of airway constriction. To illustrate the lead-up to this synergy at the cell-signaling level, pathways mediated by TGF-β, IL-4/IL-13, and NB-κB will be explored in detail. It is important to note that dozens more pathways are involved in asthmatic function, but only these three will be discussed for the purpose of conciseness.
Figure 1. Generalized intercellular signaling pathways of asthma. Note that the signaling molecules listed on the diagram encompass only a portion of the entire asthma signaling molecules repertoire. Adapted from [2].
TGF-β Signaling
Transforming Growth Factor Beta is a superfamily of cytokines that are involved in the regulation of cell growth, proliferation, differentiation, and apoptosis. They are released by a variety of cells such as lymphocytes, epithelial cells, fibroblasts, and mast cells. Due in part to its far-reaching nature, the various signaling pathways in which TGF-β participate can be subject to dysfunction and result in diseases such as cancer.
In asthmatic individuals, epithelial expression of TGF-β is upregulated, resulting in the promotion of both cell growth and cell death pathways in the lung airway cells (I.e. epithelial cells, fibroblasts, myofibroblasts, airway smooth muscle cells, endothelial cells) [3],[4]. This contributes to the perpetual damage and repair state of airway cells.
Figure 2 displays of some of the pathways which TGF-β regulates. The JNK-dependent and P38 MAPK pathways lead to cell proliferation. Conversely, TGF-β can activate Smad 7, which goes on to signal the P38 MAPK pathway for apoptosis instead. TGF-β also can stimulate the production of LTC4 Synthase, an enzyme that catalyzes the production of LTC4; in turn, LTC4 (an inflammatory mediator) can signal upregulation of TGF-β as well as stimulate constriction of airway smooth muscle cells.
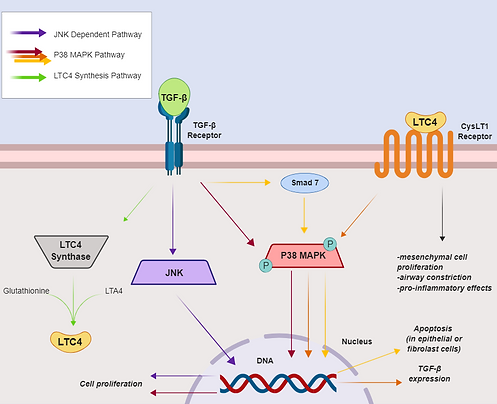
Figure 2. Effects of TGF-β signaling on lung epithelial/ mesenchymal/ fibroblast cells. Adapted from [3].
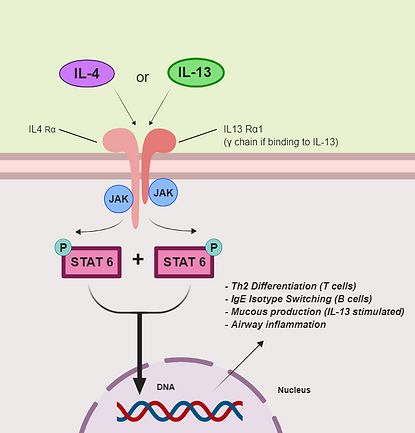
Figure 3. IL-4/IL-13 signaling pathway and effects on asthma. Adapted from [5], [7]
IL-4/IL-13 Signaling
Interleukins 4 and 13 are cytokines commonly involved in atopic immune response [5], and almost all lymphocytes can produce and release either interleukin. They are primarily responsible for the differentiation T-helper 2 cells from naïve T cells and IgE isotype switching in B cells.
High Th2 cell presence in the lungs is a major feature in asthmatic lungs [6]. The IgE produced by B cells bind to mast cells, inducing the release of more cytokines, including IL-4/IL-13; the histamine released from mast cells also lead to the release of more IL-4 and IL-13 from other lymphocytes (see Figure 1).
In the IL-4/IL-13 signaling pathway (Figure 3), either interleukin binds to the respective heterodimeric interleukin receptor complex. JAK proteins go on to phosphorylate the receptor, followed by STAT6 binding to the newly added phosphates. The STAT6 proteins are then phosphorylated by the JAK proteins, dissociate from the receptor, and form a dimer molecule which enters the nucleus. The STAT6 dimer goes on to induce Th2 cell differentiation and IgE isotype switching.
NF-κB Signaling
Nuclear Factor – κB is a homodimeric transcription factor found in numerous inflammatory signaling pathways and is found in nearly all cell types. It both induces and gets activated by many inflammatory signaling molecules [8], [9]. IκB inhibits downstream signaling of NF-κB by forming a complex with it.
It is proposed that NF-κB is continuously activated in individuals with severe asthma [8], factoring into the hyper reactive nature of asthmatic lungs. It induces transcription of inflammatory cytokines such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-8, which in turn activate cytotoxic eosinophils during inflammatory response.
In the NF-κB signaling pathway (figure 4), a signaling molecule (e.g. cytokine) would activate a Toll-like receptor or cytokine receptor found on an epithelial cell. This sends a downstream signal to activate IκB kinase (IKK); IKK goes on to phosphorylate the IκB-NF-κB complex. This allows NF-κB to dissociate and travel to the nucleus, resulting in the expression of inflammatory signals.
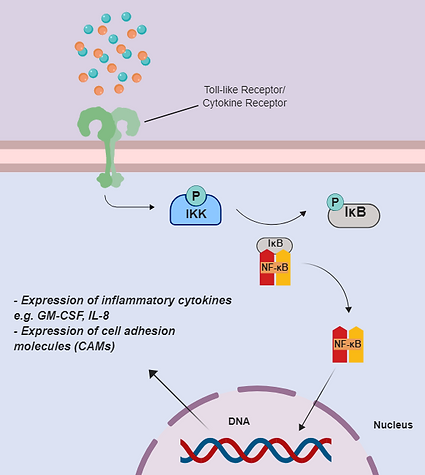
Figure 4. NF-κB signaling pathway. Adapted from [9].
Target Treatments – Signaling Pathway Intervention
As the relationships between signaling molecules and their pathways become more defined, potential targets for therapy development are revealed. Asthma was originally believed to be spasms of airway smooth muscle cells, so bronchodilator therapies were developed. These therapies included agents such as theophylline and selective β2-adrenoreceptor agonists – in essence, asthmatic patients were prescribed muscle relaxants to use as their primary treatment during an asthmatic event [1]. Later on, when asthma was redefined as an inflammatory disorder, treatment development focus was shifted onto mediation of inflammatory signaling molecules. Corticosteroid drugs proved to be effective in the downregulation of cytokines and other inflammatory proteins. [10]. Current asthma treatments are delivered as a combination of corticosteroids and long-acting B2-adrenoreceptor agonists.
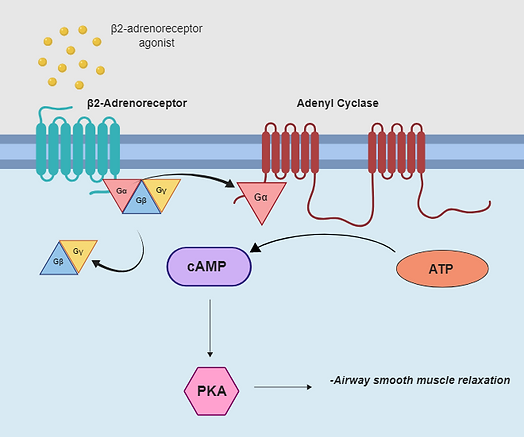
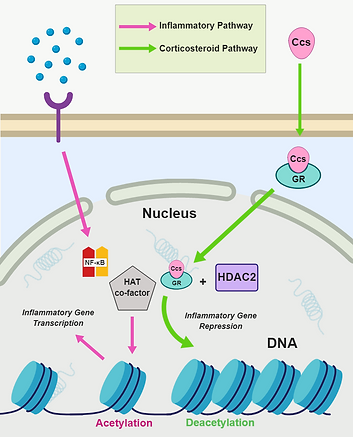
Figure 5. β2-adrenoreceptor agonist signaling pathway in airway smooth muscle. Adapted from [11].
Figure 6. Corticosteroid signaling pathway. Adapted from [10].
β2-Adrenoreceptor Agonists
β2-adrenoreceptor (β2-AR) agonist are G-protein Coupled Receptor (GCPR) ligands that bind to β-adrenergic receptors; specifically, their binding to β2-AR on an airway smooth muscle cells aid in airway relaxation [11].
The β2-adrenoreceptor agonist induces conformational change of the β2-adrenoreceptor, allowing for the hydrolyzation and dissociation of the G-protein-α-subunit from the G-protein heterotrimer. The G-protein-α-subunit goes on to bind to adenyl cyclase, which activates the formation of cyclic AMP (cAMP) and stimulates a protein kinase signaling cascade that results in the relaxation of smooth airway muscles (Figure 5).
Common β2-AR agonists
-
salbutamol (albuterol sulfate)
-
salmeterol
-
formoterol
-
vilanterol
Side Effects
Selectivity for receptors is important for the use of β2-AR agonists [11]. For example, cross-activation of β1-adrenoreceptors can lead to:
-
anxiety
-
tachycardia
-
tremors
-
sweating
Corticosteroids
Corticosteroids (AKA glucocorticoids, glucocorticosteroids, or just simply steroids) are steroid hormones that are used in the suppression of inflammatory gene expression [10]. Due to their nature as steroids, corticosteroids are non-polar/fat soluble, so they can enter cells through the lipid bilayers without the need for a protein receptor or channel.
After activation from downstream signaling (see Figure 4), NF-κB would bind to both κB recognition sites on the DNA chromatin and cofactors with intrinsic histone acetyltransferase activity (HAT). Acetylation of histone proteins allows for the unwinding of the chromatin, allowing for the transcription and expression of select inflammatory genes. Corticosteroids (Ccs) would undo the histone acetylation by entering the cell and binding to a glucocorticoid receptor (GR). The activated GR would translocate to the nucleus, recruit cofactor histone deacetylase-2 (HDAC2), and bind to HAT cofactors to inhibit and/or reverse histone acetylation. The chromatin rewinds onto the histones and inflammatory gene expression is suppressed (Figure 6).
Local Side Effects
-
dysphonia (hoarseness of voice)
-
oropharyngeal candidiasis (yeast infection)
-
cough
Systemic Side Effects
-
Adrenal suppression
-
Metabolic abnormalities
-
Growth suppression
Common Corticosteroids
-
Fluticasone
-
Budesonide
-
Beclomethasone
References
-
Holgate, S. T. (2010). A brief history of asthma and its mechanisms to modernconcepts of disease pathogenesis. Allergy Asthma Immunol Res., 2(3), 165-171
-
Pathophysiology of asthma. (2000). Int Arch Allergy Immunol, 121(suppl 1), 25-31.
-
Makinde, T., Murphy, R. F., & Agrawal, D. K. (2007). The regulatory role of TGF- B in airway remodelingin asthma. Immunology and Cell Biology, 85, 348-356.
-
Semlali, A., & et al. (2008). TGF- B suppresses EGF-induced MAPK signaling andproliferation in asthmatic epithelial cells. Am Respir Cell Mol Biol, 38, 202-208.
-
Kabesch, M., & et al. (2005). IL-4/IL-13 pathway genetics strongly influencserum IgE levels and childhood asthma. J Allergy Clin Immunol, 117(2), 269-274.
-
Packard, K. A., & Khan, M. M. (2002). Effects of histamine on Th1/Th2 cytokine balance. International Immunopharmacology, 3, 909-920.
-
Erle, D. J., & Sheppard, D. (2014). The cell biology of asthma. J. Cell Biol., 205(5), 621-631.
-
Gagliardo, R., & et al. (2003). Persistent activation of nuclear Factor–kB signalingpathway in severe uncontrolled asthma. Am J Respir, 168, 1190-1198.
-
Schuliga, M. (2015). NF-kappaB signaling in chronic inflammatory airway disease. Biomolecules, 5, 1266-1283.
-
Barnes, P. J. (2010). Inhaled corticosteroids. Pharmaceuticals (Basel), 3(3), 514-540.
-
Billington, C. K., Penn, R. B., & Hall, I. P. (2017). Β2-agonists. Handb Exp Pharmacol, 237, 40
*All images were made using BioRender.